![]()


SMA(愐悜惈嬝堔弅徢)偲偼
娔廋丂搶嫗彈巕堛壢戝妛晅懏堚揱巕堛椕僙儞僞乕丂嵵摗壛戙巕愭惗
SMA偵偮偄偰丄俻仌俙宍幃偱夝愢偟傑偟傚偆丅
俻丗SMA偲偼偳傫側昦婥偱偡偐丠
俻丗SMA偺奺宆偺徢忬偵偮偄偰丄傕偆彮偟徻偟偔嫵偊偰壓偝偄丅
俻丗偲偙傠偱丄戝恖偺曽偱僂僃儖僪僯僢僸丒儂僼儅儞(Werdnig-Hoffmann)昦偲恌抐偝傟偰偄傞曽傕偄傑偡偑乨乨乨
俻丗SMA偼偳偺傛偆偵恌抐傪偡傞偺偱偡偐丠
俻丗SMA偼堚揱昦側偺偱偡偐丠
俻丗堚揱傗堚揱巕曄堎偲偼偳偆偄偆偙偲側偺偱偡偐丠
俻丗SMA偺堚揱巕偵偮偄偰丄傕偆彮偟徻偟偔愢柧偟偰偔偩偝偄丅
俻丗SMA偱偼堚揱巕偼偳偺傛偆偵側偭偰偄傞偺偱偡偐丠
俻丗SMA偺堚揱巕恌抐偼偳偺傛偆偵峴偆偺偱偡偐丠
俻丗SMA偺帯椕偵偮偄偰愢柧偟偰壓偝偄丅
俻丗SMA偺帯椕栻偼丠
俻丗SMA偲ALS乮嬝堔弅惈懁嶕峝壔徢乯偺堘偄偼丠
俻丗SMA偲媴愐悜惈嬝堔弅徢偼摨偠昦婥偱偡偐丠
俻丗恄宱撪壢偱愐悜惈恑峴惈嬝堔弅徢偲恌抐偝傟傑偟偨偑乨乨乨
俻丗SMA偲偼偳傫側昦婥偱偡偐丠
俙丗SMA(Spinal Muscular Atrophy)偲偼丄愐悜偺塣摦恄宱嵶朎(愐悜慜妏嵶朎)偺昦曄偵傛偭偰婲偙傞恄宱尨惈偺嬝堔弅徢偱丄ALS乮嬝堔弅惈懁嶕峝壔徢乯偲摨偠塣摦僯儏乕儘儞昦偺斖醗偵擖傞昦婥偱偡丅峀媊偺SMA偲偼恾侾偺傛偆偵丄條乆側尨場傪傕偮徢岓孮偲峫偊傜傟傑偡偑丄偄偢傟傕懱姴傗巐巿偺嬤埵晹偵桪埵偺嬝偺扙椡丄嬝堔弅傪帵偟傑偡丅堦曽丄嫹媊偺SMA偼戞俆愼怓懱偵昦場堚揱巕傪帩偮楎惈堚揱昦偱丄敪徢擭楊偲廳徢搙偵傛偭偰3宆偵暘椶偝傟傑偡丅1)
丂崙嵺揑偵偼嫹媊偺SMA傪丄堦斒揑偵SMA偲徧偟偰偄傑偡丅崙嵺暘椶偱偼SMA傪峀媊丄嫹媊偲暘偗偰偄側偄偙偲偐傜丄尰嵼偺偲偙傠丄峀媊丄嫹媊偲偄偆暘椶偼偁偔傑偱傕杮夛偺峫偊曽偱偁傞偙偲傪偍抐傝偟偰偍偒傑偡丅
- 1宆丄廳徢宆丄媫惈擕帣宆丄僂僃儖僪僯僢僸丒儂僼儅儞(Werdnig-Hoffmann)昦
丂敪徢偼惗屻6儢寧傑偱丅惗奤嵖埵曐帩晄壜擻偱偡丅捠忢偺堛妛彂偺婰弎偵傛傟偽丄2嵨傑偱偵朣偔側傞偲偝傟偰偄傑偡偑丄偦傟偼恖岺屇媧婍傪巊梡偟側偄応崌偱偡丅揔愗側恖岺屇媧娗棟傪庴偗偰偄傞恖偺拞偵偼丄惉恖偵払偟偰偄傞恖傕偄傑偡丅傑偨丄SMA2宆傗3宆偺恖偺拞偵偼丄擕帣抜奒偱1宆乮僂僃儖僪僯僢僸丒儂僼儅儞昦乯偱偁傞偲偄偆妋掕恌抐傪庴偗丄屻偵側偭偰偐傜2宆傕偟偔偼3宆偱偁傞偲尵傢傟偰偄傞働乕僗傕偁傝傑偡丅
- 2宆丄拞娫宆丄枬惈擕帣宆
丂敪徢偼1嵨6儢寧傑偱丅嵖埵曐帩偼壜擻偱偡偑丄惗奤丄婲棫傗曕峴偼晄壜擻偱偡丅擕帣婜憗婜偵朣偔側傞偙偲偼偁傝傑偣傫丅愩偺慄堐懇惈澒弅傗堔弅丄庤巜偺怳愴偑傒傜傟傑偡丅銯斀幩偼尭庛傑偨偼徚幐丅師戞偵懁淽偑挊柧偵側傞偨傔丄偦傟傪梊杊偡傞偨傔偺憗婜夘擖乮儕僴價儕乯偑昁梫偲側傝傑偡丅
- 3宆丄寉徢宆丄枬惈宆丄僋乕僎儖儀儖僌丒償僃儔儞僟乕(Kugelberg-Welander)昦
丂敪徢偼1嵨6儢寧埲崀丅帺棫曕峴傪妉摼偟傑偡偑丄師戞偵揮傃傗偡偄丄曕偗側偄丄棫偰側偄偲偄偆徢忬偑偱偰偒傑偡丅屻偵丄忋巿偺嫇忋傕崲擄偵側傝傑偡丅
俻丗SMA偺奺宆偺徢忬偵偮偄偰丄傕偆彮偟徻偟偔嫵偊偰壓偝偄丅
俙丗1宆偼嬝椡掅壓偑廳徢偱慡恎惈偱偡丅擠怭拞偺戀摦偑庛偄椺偼戀撪偐傜偺敪徢傪峫偊偝偣傑偡丅敪徢屻丄塣摦敪払偼掆巭偟丄懱姴傪摦偐偡偙偲傕偱偒偢丄嬝嬞挘傗嬝椡偑掅壓偟偰偄傞丄偄傢備傞僼儘僢僺乕僀儞僼傽儞僩偺忬懺傪帵偟傑偡丅巟偊側偟偵嵗傞偙偲偑偱偒偢丄歁擕崲擄丄殝壓崲擄丄岆殝丄屇媧晄慡傪敽偄傑偡丅愩偺慄堐懇惈澒弅偑傒傜傟丄怺晹銯斀幩偼徚幐偟偰偄傑偡丅恖岺屇媧婍傪梡偄側偄応崌丄巰朣擭楊偼暯嬒6乣9僇寧丄95亾偼18僇寧傑偱偵巰朣偡傞偲偄傢傟偰偍傝丄惗柦傪媬偆偨傔偵偼丄婥娗撪憓娗傗婥娗愗奐偲恖岺屇媧娗棟偑昁梫偱偡丅巆擮側偑傜擔杮偱偼怤廝揑側婥娗愗奐偵傛傞恖岺屇媧娗棟偑庡棳偱偡偑丄旕怤廝揑側恖岺屇媧乮NIPPV乯偱彆偗傜傟傞壜擻惈偼丄擔杮偱傕嬤偄彨棃偵弌偰偔傞傕偺偲婜懸偝傟偰偄傑偡丅傑偨堦曽偱丄屆偔偐傜SMA偺椪彴壠偺娤嶡偵傕偄傢傟偰偄傞偙偲偼丄1宆偺揟宆椺偲峫偊傜傟偰偄傞徢椺偱丄屇媧婍側偟偵宱夁傪偟偰偄傞恖偑偄傞偙偲偱偡丅悽奅揑側屇媧娗棟偺尃埿幰偱偁傞Bach嫵庼偵傛傟偽丄乽僞僀僾1偺彫帣偺椉恊偼丄捠忢丄姵帣偑1擭偐傜2擭埲撪偵朣偔側傞偱偁傠偆 偲偄傢傟傞偑丄3偐寧埲撪偵敪徢偟偨懡偔偺徢椺偱偼丄夘彆側偟偵怘帠偑偱偒丄帺敪屇媧偑偁傝丄20嵨偺帪揰偱丄敪徢偺抶偄姵帣傛傝傕嫮偄偙偲偑偁傞丅揟宆揑側僞僀僾1偺彫帣偑惉恖傑偱惗懚偟摼傞乿乮僕儑儞丒僶僢僋亀恄宱嬝幘姵偺昡壙偲儅僱僕儊儞僩亁恌抐偲帯椕幮丄1999擭丄17-18暸乯丅
丂2宆偼巟偊側偟偵棫偭偨傝丄曕偄偨傝偡傞偙偲偑偱偒傑偣傫丅愩偺慄堐懇惈澒弅傗堔弅丄庤巜偺怳愴偑傒傜傟傑偡丅銯斀幩偼尭庛傑偨偼徚幐丅惉挿偲偲傕偵懁淽偑挊柧偵側傝傑偡丅2宆偺巕偳傕払偵偍偄偰偼丄娭愡峉弅偲懁淽偺梊杊偑儕僴價儕僥乕僔儑儞偺栚揑偱偡丅傑偨丄忋婥摴姶愼偵堷偒懕偄偰丄攛墛傗柍婥攛偵側傝丄屇媧晄慡偵娮傞偙偲偑偁傝傑偡丅晛抜偐傜偺攔醾偺孭楙偲姶愼帪偺揔愗側堛椕懳墳偑昁梫偱偡丅SMA偺巕偳傕払偺抦擻偑桪傟偰偄傞偙偲偑丄SMA偺屆揟偱偁傞Hoffmann偺榑暥偵傕婰嵹偝傟偰偄傑偡丅SMA偺巕偳傕偨偪偺SMA偵傛傞恎懱揑側妶摦偺惂尷傪忣曬婡婍傗偝傑偞傑側暉巸婡婍偵傛偭偰丄僇僶乕偱偒傞偙偲偑朷傑傟傑偡丅
丂3宆偱偼棫偭偨傝曕偄偨傝偱偒偰偄偨偺偵丄揮傃傗偡偄丄曕偗側偄丄棫偰側偄偲偄偆徢忬偑偱偰偒傑偡丅師戞偵丄忋巿偺嫇忋傕崲擄偵側傝傑偡丅敪徢偺帪婜偼梒帣婜丄彫帣婜偺偙偲傕偁傝傑偡偑丄惉恖偺敪徢傕懡偔尒傜傟傑偡丅
丂惉恖偺敪徢偺宆傪4宆偲屇傇偙偲傕偁傝傑偡丅惉恖偺敪徢偺応崌偼懁淽偼尒傜傟傑偣傫偑丄彫帣婜埲慜偺敪徢偱偼懁淽偑惗偠傑偡丅敪徢擭楊偑抶偄傎偳恑峴偺僗僺乕僪偼娚傗偐偱丄梊屻椙岲偲弎傋傜傟偰偄傑偡丅壓埵塣摦僯儏乕儘儞偺傒偺忈奞偱偁傝丄嬝堔弅惈懁嶕峝壔徢(ALS)偑忋埵僯儏乕儘儞傕忈奞偝傟傞偺偲斾妑偝傟傑偡丅乮俻丗SMA偲ALS乮嬝堔弅惈懁嶕峝壔徢乯偺堘偄偼丠 嶲徠乯
俻丗偲偙傠偱丄戝恖偺曽偱僂僃儖僪僯僢僸丒儂僼儅儞(Werdnig-Hoffmann)昦偲恌抐偝傟偰偄傞曽傕偄傑偡偑乨乨乨
俙丗SMA偺奺宆偺暘椶偼挿偄偙偲丄恌抐偡傞堛巘偵傛偭偰摑堦偝傟偨傕偺偱偼偁傝傑偣傫偱偟偨丅1991擭崰偐傜丄尨場偺堚揱巕傪敪尒偡傞偨傔偵柧妋側暘椶偲恌抐婎弨傪妋棫偡傞偙偲偑昁梫偱偁傞偲偄偆峫偊偺傕偲偵丄崙嵺SMA嫤夛(International SMA Consortium)偑慻怐偝傟偰丄忋婰偺暘椶偑崌堄偝傟傑偟偨丅偟偐偟丄傑偩SMA亖僂僃儖僪僯僢僸丒儂僼儅儞昦偲偄偆恌抐傪偟偰偄傞堛巘傕偄傞傛偆偱偡丅傑偨丄尰嵼偺暘椶偱傕1宆丄2宆丄3宆偺偦傟偧傟偺嫬奅偼丄慄偱暘偗傜傟傑偣傫丅
俻丗SMA偼偳偺傛偆偵恌抐傪偡傞偺偱偡偐丠
俙丗SMA偵偼丄師偺傛偆側恌抐婎弨2)偑偁傝丄嬝椡掅壓偲扙恄宱偺強尒偵傛傝椪彴揑偵恌抐偟傑偡丅傑偨嵟嬤偼屻偵弎傋傞傛偆側堚揱巕恌抐偵傛偭偰丄懡偔偺椺偱妋掕恌抐偑偱偒傞傛偆偵側偭偰偒傑偟偨丅俻丗SMA偼堚揱昦側偺偱偡偐丠
- 嬝椡掅壓偺強尒
亅嵍塃懳徧惈偺忈奞
亅墦埵嬝(庤巜傗懌愭偺嬝擏)傛傝嬤埵嬝(傛傝懱姴偵嬤偄嬝擏)偺忈奞偑廳偄
亅忋巿傛傝壓巿偺忈奞偑廳偄
亅嬰姴偍傛傃巐巿偲傕偵忈奞偝傟傞
- 扙恄宱偺強尒
亅愩偺慄堐懇惈澒弅(愩偺嵶偐偄恔偊)丄庤巜偺怳愴
亅嬝惗専偱堔弅嬝慄堐偺廤抍偲1宆慄堐偺旍戝
亅嬝揹恾偱恄宱尨惈曄壔
- 彍奜恌抐丂埲壓偺傛偆側強尒偑偁傞応崌偼SMA偲偼峫偊偵偔偄偱偡丅
亅拞悤恄宱婡擻忈奞
亅娭愡峉弅徢
亅奜娽嬝丄墶妘枌丄怱嬝偺忈奞
亅挳妎忈奞
亅挊偟偄婄柺嬝滊姵
亅抦妎忈奞
亅寣惔僋儗傾僠儞僉僫乕僛(CK)抣偑惓忢忋尷偺10攞埲忋
亅塣摦恄宱揱摫懍搙偑惓忢壓尷偺70亾埲壓
亅抦妎恄宱妶摦揹埵偺堎忢
俙丗嫹媊偺SMA偼忢愼怓懱惈楎惈堚揱偺昦婥偱偡丅儈僋儘偺儗儀儖偱愢柧傪偟傑偟傚偆丅僸僩偺嵶朎偺拞偵偼妀偑偁傝傑偡丅偙偺妀偵偼晝恊桼棃丄曣恊桼棃偺2杮1慻偺愼怓懱偑慡晹偱46杮丄23慻偁傝傑偡丅1慻偑惈愼怓懱偱丄巆傝偺22慻偑忢愼怓懱偱偡丅忢愼怓懱偵偼1斣偐傜22斣傑偱斣崋偑偮偄偰偄傑偡丅偙偺偆偪5斣偺愼怓懱偺挿榬偺13偲偄偆応強(5q13)偵SMA偺堚揱巕(SMA堚揱巕)偑懚嵼偡傞偙偲偑傢偐傝傑偟偨丅1丄2丄3宆偲傕丄偙偺SMA堚揱巕偺曄堎偵傛偭偰惗偠傞偺偱偡丅晝恊桼棃偺SMA堚揱巕偲曣恊桼棃偺SMA堚揱巕偑嫟偵曄堎傪帵偟偰偄傞応崌偵丄偦偺巕偼SMA偵側傝傑偡丅晝恊桼棃傑偨偼曣恊桼棃偺堚揱巕偑偳偪傜偐1偮偩偗曄堎偟偰偄傞応崌偼慡偔柍徢忬偱偁傝丄偙偺応崌傪曐場幰偲偄偄傑偡丅曐場幰偼慡偔徢忬偼偁傝傑偣傫丅曐場幰摨巑偺寢崶偺応崌丄偍巕偝傫偑偙偺昦婥偵側傞壜擻惈偼1/4 (25亾)偱偡丅1宆偺曐場幰偺昿搙偼墷暷偱偼60乣80恖偵1恖丄2宆丄3宆偼76乣111恖偵1恖丄偲偄傢傟偰偄傑偡偑丄擔杮偱偼墷暷傛傝彮側偄傛偆偱偡丅曐場幰偺昿搙傪100恖偵1恖偲壖掕偡傞偲丄曐場幰摨巑偺寢崶偼1/100亊1/100亖1/10,000偱偁傝丄偍巕偝傫偑SMA偲側傞壜擻惈偼1/10,000亊1/4亖1/40,000偲側傝傑偡丅廬偭偰丄姵幰偝傫屼杮恖傗偦偺孼掜巓枀偑寢崶偝傟偰傕丄偄偲偙摨巑側偳偺寣懓寢崶偱側偗傟偽丄偍巕偝傫偑SMA偵側傞壜擻惈偼傎偲傫偳偁傝傑偣傫丅
俻丗堚揱傗堚揱巕曄堎偲偼偳偆偄偆偙偲側偺偱偡偐丠
俙丗僸僩偺僎僲儉僾儘僕僃僋僩偑恑傒丄懡偔偺堚揱巕偑暘偐傞傛偆側帪戙偵側偭偰偒傑偟偨丅僸僩偺堚揱巕偼慡晹偱栺係枩屄偁傞偲悇掕偝傟偰偄傑偡丅堚揱巕偺尋媶傪偟偰偄傞偲丄椙偔暘偐偭偰偔傞偙偲側偺偱偡偑丄堚揱巕傪宍嶌傞DNA偲偼A乮傾僨僯儞乯丄 T乮僠儈儞乯丄 C乮僔僩僔儞乯丄 G乮僌傾僯儞乯偺係偮偺墫婎偐傜惉偭偰偍傝丄堚揱巕曄堎偲偼丄偦偺墫婎攝楍偑椺偊偽丄ATCG偺偼偢偑丄ATGG偵側偭偨傝丄ATT偵側偭偨傝偟偰偄傞乽偩偗乿偺偙偲側偺偱偡丅恊偐傜DNA忣曬傪傕傜偆応崌偵丄傑偨帺暘偺嵶朎偑DNA忣曬偵傛偭偰憹偊傞応崌偵丄扤偵偍偄偰傕丄60壄偺DNA偲4枩偺堚揱巕偑撉傒娫堘偄傪婲偙偝側偄偼偢偼側偄偺偱偡丅夝愅偺恑揥偵傛偭偰丄僸僩偼扤偱傕丄偦傟傜偺堚揱巕偵悢屄偺曄堎傪帩偭偰偄傞偲峫偊傜傟傞傛偆偵側偭偰偒傑偟偨丅偮傑傝丄巹偨偪偼傒傫側丄徢忬偼側偔偰傕壗傜偐偺昦婥偺堚揱巕曄堎傪帩偭偰偄偰丄曐場幰偱偁傞偲偄偊傑偡丅楎惈堚揱偺偨傔乽嬼慠乿昦婥偑敪徢偟偰偄側偄偩偗偩偲傕尵偆偙偲偑偱偒傞偺偱偡丅椺偊偽SMA偲偄偆昦婥偼丄乽嬼慠乿偟偐傕乽偩傟偺愑擟偱傕側偔乿偦偺傛偆側堚揱巕曄堎偑SMN堚揱巕偵惗偠偰偄傞偙偲偵傛偭偰婲偙傞偺偱偡丅
俻丗SMA偺堚揱巕偵偮偄偰丄傕偆彮偟徻偟偔愢柧偟偰偔偩偝偄丅
俙丗1995擭偵SMA偺尨場堚揱巕(SMA堚揱巕)偱偼側偄偐偲峫偊傜傟傞堚揱巕偑2偮敪尒偝傟傑偟偨丅塣摦恄宱嵶朎惗懚(survival motor neuron:棯偟偰SMN)堚揱巕3)偲恄宱嵶朎傾億僩乕僔僗梷惂抈敀(neuronal apoptosis inhibitory protein:棯偟偰NAIP)堚揱巕4)偱偡丅偙偺2偮偺堚揱巕偼愼怓懱5q13偵偍偄偰旕忢偵愙嬤偟偰懚嵼偟偰偄傑偡丅偙偺2偮偺堚揱巕偺偆偪丄SMN堚揱巕偺曽偑SMA偺尨場堚揱巕偱偁傞偲峫偊傜傟偰偒偰偄傑偡丅偙偺SMN堚揱巕偺僙儞僩儘儊傾懁偵偼丄SMNcopy堚揱巕偲柤晅偗傜傟偨丄SMN堚揱巕偲偼5墫婎懳偺傒偑堎側偭偰偄傞堚揱巕偑懚嵼偟偰偄傑偡丅廬偭偰SMN堚揱巕偺偙偲傪SMNsma堚揱巕偲偐丄僥儘儊傾懁偵偁傞偺偱SMNtel堚揱巕偲尵偭偰偄傑偡丅SMNtel堚揱巕傗NAIP堚揱巕偑巌偭偰偄傞抈敀幙偺摥偒偼柧傜偐偵偝傟偮偮偁傝傑偡丅SMNcopy堚揱巕偵偮偄偰偼偦偺摥偒偼晄柧偱偡丅SMN抈敀偼嵶朎偺妀偵懚嵼偟丄RNA寢崌抈敀偲斀墳偡傞傕偺偱偁傠偆偲峫偊傜傟偰偄傑偡丅NAIP偼崺拵嵶朎偺傾億僩乕僔僗傪梷惂偡傞抈敀幙偲峔憿偑帡偰偄傞偨傔丄恄宱嵶朎偺傾億僩乕僔僗傪梷惂偡傞抈敀幙偲峫偊傜傟偰偄傑偡丅
俻丗SMA偱偼堚揱巕偼偳偺傛偆偵側偭偰偄傞偺偱偡偐丠
俙丗SMNtel堚揱巕傗NAIP堚揱巕偑寚懝偟偰偄傑偡(寚幐偲偄偄傑偡)丅慡偰偺姵幰偝傫偱寚幐偑傒傜傟傞偺偱偼側偔丄SMNtel堚揱巕偼SMA偺姵幰偝傫偺栺87亾丄NAIP堚揱巕偱偼栺17亾偵寚幐傪擣傔傑偡丅墷暷偱偼寚幐傪帵偡椺偑傢偑崙傛傝懡偔丄偦傟偧傟95亾丄60亾傎偳偱偡丅1宆偱偼SMNtel堚揱巕偲NAIP堚揱巕偺寚幐傪擣傔丄廳徢椺偱偼寚幐偑戝偒偄孹岦偑偁傞偙偲傪帵嵈偟偰偄傑偡丅廬偭偰丄杮徢偺妋掕恌抐偵SMNtel堚揱巕丄NAIP堚揱巕偺寚幐偼桳岠偱偡偑丄寚幐傪帵偝側偄椺傕偁傞偙偲偵棷堄偟偰偔偩偝偄丅
俻丗SMA偺堚揱巕恌抐偼偳偺傛偆偵峴偆偺偱偡偐丠
俙丗堚揱巕恌抐偵偼捈愙揑恌抐偲娫愙揑恌抐偑偁傝傑偡丅偄偢傟偺応崌傕丄DNA偼懱偺偳偺嵶朎偐傜嵦偭偰傕傛偄偺偱偡丅堦斒偵偼5-10ml傎偳偺嵦寣傪峴偄傑偡丅嬝惗専傪峴偭偰偄傞応崌偼丄偦偺嬝擏偐傜DNA傪嵦傞偙偲傕壜擻偱偡丅捈愙揑恌抐偼慜偵弎傋偨SMN丄NAIP椉堚揱巕偺寚幐傪挷傋傞曽朄偱偡丅娫愙揑恌抐偼丄DNA偺懡宆偑恊偐傜巕嫙偵偳偺傛偆偵揱傢偭偰偄傞偐傪傒傞曽朄偱偡丅SMN堚揱巕嬤朤偺儅僀僋儘僒僥儔僀僩DNA懡宆儅乕僇乕傪梡偄偰丄CA孞傝曉偟攝楍偺僒僀僘傪挷傋傑偡丅姵幰偝傫偼晝恊偺堚揱巕宆偲曣恊偺堚揱巕宆傪侾杮偢偮傕傜偭偰偄傑偡丅姵幰偝傫偺孼掜偵偍偄偰丄晝曽偺堚揱巕宆偑寬忢偱偡偑丄曣曽偺堚揱巕宆偑姵幰偝傫偲摨條偱偁傞応崌(傑偨偼媡偵丄晝曽偺堚揱巕宆偑姵幰偝傫偲摨條偱偡偑丄曣曽偺堚揱巕宆偑寬忢偱偁傞応崌)丄曐場幰偲敾掕偟傑偡丅
俻丗SMA偺帯椕偵偮偄偰愢柧偟偰壓偝偄丅
俙丗杮徢偱偼崻杮帯椕偼偄傑偩妋棫偟偰偄傑偣傫丅SMA偺1宆傗2宆偺擕帣婜偵敪徢偡傞姵幰偝傫偱偼丄庼擕傗殝壓偑崲擄側偨傔僠儏乕僽塰梴偑昁梫側偲偒偑偁傝傑偡丅傑偨丄屇媧婍姶愼丄柍婥攛傪孞傝曉偡徢椺偑懡偔丄偙傟偑梊屻傪戝偒偔嵍塃偟傑偡丅旲儅僗僋恖岺屇媧朄(NIPPV)偼桳岠偲峫偊傜傟傑偡偑丄擕帣椺傊偺巊梡偵偼懡偔偺崲擄傪敽偄傑偡丅傑偨丄嬝椡偵偁傢偣偨塣摦孭楙丄娭愡峉弅偺梊杊側偳偺儕僴價儕僥乕僔儑儞偑昁梫偱偡丅3宆偱偼曕峴壜擻側忬懺傪側傞傋偔挿婜偵堐帩偱偒傞傛偆偵丄傑偨娭愡峉弅偺梊杊偺偨傔偵傕丄儕僴價儕僥乕僔儑儞傪峴偄丄憰嬶偺巊梡側偳傪専摙偟丄彫帣恄宱堛丄恄宱撪壢堛丄惍宍奜壢堛丄婡擻孭楙巑偺楢実偑昁梫偱偡丅偄偢傟偵偟偰傕丄偙傟傜偺SMA偺昦場丄昦懺偑柧傜偐偵偝傟傞偲嫟偵丄夋婜揑側帯椕朄偺奐敪偑婜懸偝傟傑偡丅
俻丗SMA偺帯椕栻偼丠
俙丗杮徢偵桳岠側帯椕栻偼傑偩奐敪偝傟偰偄傑偣傫丅嶲峫傑偱偵偄偔偮偐偺忣曬傪宖嵹偟偰偍偒傑偡丅
栻昳柤 岠壥 儕儖僥僢僋 ALS偺帯椕栻丅嵶朎儗儀儖偱僌儖僞儈儞巁傪梷惂偡傞丅SMA傊偺岠壥偑偁傞偐偼妋擣偝傟偰偍傜偢丄奀奜偱巊梡椺偁傞偑偦偺寢壥偼曬崘偝傟偰偄側偄丅擔杮偱偼SMA偺恌抐偱偼栻傪弌偣側偄丅SMA偑恑峴拞偺応崌偼僌儖僞儈儞巁偑嵶朎儗儀儖偱憹壛偟偰偄偰丄塭嬁偑偁傞偐傕偟傟側偄偺偱岠壥偺壜擻惈偼偁傞丅堦曽丄恑峴偑巭傑偭偰偄傞応崌偼僌儖僞儈儞巁偺夁忚偼峫偊傜傟偢丄儕儖僥僢僋偺岠壥偼側偄偲巚傢傟傞丅 僈僶儁儞僠儞 婎杮揑偵儕儖僥僢僋偲摨偠丅奀奜偱専摙偝傟偰偄傞偑丄傑偩寢壥偼曬崘偑弌偰偄側偄丅 僋儗傾僠儞 嬝擏峔惉惉暘偺堦庬丅傾儊儕僇戝儕乕僌偺儅僌儚僀儎慖庤偑暈梡偟偰偄傞偙偲偱桳柤丅搶嫗彈巕堛戝偱偼儈僩僐儞僪儕傾擼嬝徢偱帯椕岠壥敾掕拞丅尰嵼偺偲偙傠岠壥偑尒傜傟偰偍傝暃嶌梡傕偁傑傝側偄丅SMA偵偮偄偰偼幚巤偡傞偐専摙拞丅梫朷偑偁傟偽慜岦偒偵峫偊傞偑丄戝妛偺椣棟埾堳夛傪捠偡昁梫偑偁傞丅堦廡娫掱搙擖堾偟側偑傜岠壥偲暃嶌梡傪妋擣屻偵奜棃偱専摙偡傞偙偲偵側傞偑丄岠壥偺昡壙曽朄摍偺専摙壽戣偁傝丅 TRH 恄宱揱払暔幙丅TSH乮峛忬態巋寖儂儖儌儞乯傪擼撪偱梀棧偝偣傞儂儖儌儞丅愐悜彫擼曄惈徢傊偺岠壥偼幚徹偝傟偰偄偰曐尟揔墳偑偁傞丅SMA偵偮偄偰搶嫗彈巕堛戝偱傕巊梡偟偨宱尡偑偁傞丅00擭9寧7擔偵揷曈惢栻偐傜宱岥TRH嵻乮彜昳柤丗僙儗僕僗僩乯偑弌偨偑丄SMA偼曐尟揔墳奜偲側偭偰偄傞丅
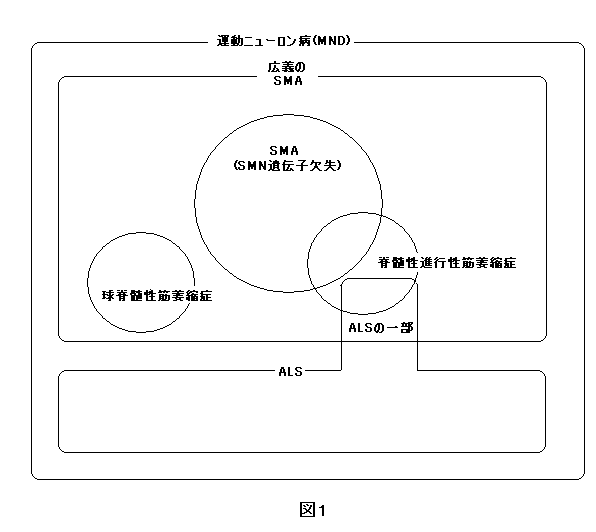
俻丗SMA偲ALS乮嬝堔弅惈懁嶕峝壔徢乯偺堘偄偼丠
俙丗SMA傕ALS傕摨偠塣摦僯儏乕儘儞偺曄惈傪庡懱偲偡傞幘姵偱偁傝丄塣摦僯儏乕儘儞昦偺斖醗偵擖傞昦婥偱偡丅
丂 SMA ALS 堚揱惈 嫹媊偺SMA偼忢愼怓懱惈楎惈堚揱 堦晹偼忢愼怓懱惈桪惈堚揱乮偦偺懠偼屒敪椺乯 昦曄晹埵 庡偵愐悜慜妏嵶朎丄壓埵塣摦僯儏乕儘儞偵塭嬁 壓埵塣摦僯儏乕儘儞偩偗偱側偔忋埵塣摦僯儏乕儘儞傊傕塭嬁 銯斀幩 庛偄 昦弶婜偵槾恑乮夁搙偵斀幩乯 敪徢帪婜 0嵨偐傜惉恖傑偱 憗偔偰傕10戙埲崀 SMA偱傕愐悜懁嶕丄愐悜屻嶕乮抦妎偵塭嬁乯偵昦曄偑偁傞偙偲傕偁傞丅
SMA偲SPMA偼摨偠昦婥傪巜偡丅SPMA偺P偼Progressive乮恑峴惈乯偺堄枴偩偑嵟嬤偼徣偔偙偲偑懡偄丅尰嵼偱傕堛巘偵傛偭偰偼P傪偮偗傞応崌傕偁傞偑崙嵺揑偵偼SMA偲徧偝傟偰偄傞丅
俻丗媴愐悜惈嬝堔弅徢偼SMA偲摨偠昦婥偱偡偐丠
俙丗媴愐悜惈嬝堔弅徢偼峀媊偺SMA偵擖傝傑偡丅働僱僨傿乕丒傾儖僞乕丒僒儞僋乮Kennedy-Alter-Sung)昦偲傕偄傢傟傞塣摦僯儏乕儘儞昦偱偡丅SMA偺徢忬偵丄媴徢忬偲偄偭偰暔傪堸傒崬傓婡擻偺忈奞傪崌暪偟傑偡丅偦偺摿挜偼埲壓偺捠傝偱偡丅丂働僱僨傿乕昦偼嶰墫婎斀暅攝楍偑墑挿偡傞偙偲偑堚揱巕儗儀儖偺摿挜偱偡丅X愼怓懱偺挿榬偺Xq11.2-12偵埵抲偡傞傾儞僪儘僎儞儗僙僾僞乕堚揱巕偺戞1僄僋僜儞偵懚嵼偡傞CAG偺斀暅攝楍偼惓忢偱偼21乣26偱偡偑丄杮徢偱偼43乣51偵憹壛偟偰偄傑偡丅偙偺傾儞僪儘僎儞儗僙僾僞乕堚揱巕偺曄堎偵傛偭偰丄傾儞僪儘僎儞晄慡徢偲偟偰偺惈態婡擻忈奞傪帵偡偺偱偡丅CAG偺斀暅攝楍偺堎忢墑挿偑挿偄傎偳丄嬝椡掅壓偺敪徢偑憗偔丄廳徢偲側傞孹岦偑偁傝傑偡偑丄摨偠CAG偺斀暅悢偱傕丄敪徢擭楊傗廳徢搙偑堎側傞偙偲傕偁傝丄堚揱巕埲奜偺梫慺偺娭梌傕偁傞偲峫偊傜傟傑偡丅廬偭偰丄椪彴徢忬偺摿挜傕丄堚揱巕揑偵傕嫹媊偺SMA偲偼堎偭偰偄傑偡丅偟偐偟丄愐悜偵桼棃偡傞嬝堔弅徢偲偄偆揰偑嫟捠偱偡丅
- X楢嵔惈楎惈堚揱宍幃傪偲傝傑偡丅
- 20乣40嵨戙偺庡偲偟偰抝惈偵敪徢偟傑偡丅
- 巐巿嬤埵嬝偺嬝椡掅壓丄嬝堔弅丄庤巜偺怳愴丄嬝醶澒丄婄柺嬝偍傛傃愩偺堔弅偵傛傞旲惡丄峔壒忈奞偑擣傔傜傟傑偡丅傑偨丄嬝廂弅帪偺嬝慄堐懇惈澒弅偑婄柺丄愩偵娤嶡偝傟傑偡丅
- 彈惈壔擕朳丄惛憙堔弅丄彈惈梡旂晢曄壔側偳寉搙偺抝惈惈態婡擻忈奞偺崌暪傪摿挜偲偟傑偡丅
- 枛婜偵偼夌彴忬懺偵側傝丄殝壓忈奞偵傛傞岆殝惈攛墛傪婲偙偟傗偡偔側傝傑偡丅
俻丗恄宱撪壢偱愐悜惈恑峴惈嬝堔弅徢偲恌抐偝傟傑偟偨偑乨乨乨
俙丗惉恖傪懳徾偲偡傞恄宱撪壢偱偼愐悜惈恑峴惈嬝堔弅徢乮SPMA乯偲偄偆恌抐偑峴傢傟傞偙偲偑偁傝傑偡偑丄偙傟偼嫹媊偺SMA傪娷傓丄傛傝峀媊偺徢岓孮偲峫偊傜傟傑偡丅応崌偵傛偭偰偼ALS偲偺嫬奅偑晄柧妋側働乕僗傕尒傜傟傑偡丅
暥專
1) Campbell MJ, Munsat TL: Motor neurone diseases. In Disorders of voluntary muscle, ed by Walton J, Karpati G, Hilton-Jones D, Churchill Livingstone, London, 1994, pp 879-919
2) Munsat TL: Workshop report. International SMA Collaboration. Neuromusc Disord 1: 81, 1991
3) Lefebvre S, Burglen L, Reboullet S, et al: Identification and characterization of a spinal muscular atrophy - determining gene. Cell 80: 155-165, 1995
4) Roy N, Mahadevan MS, McLean M, et al: The gene for neuronal apoptosis inhibitory protein is partially deleted individuals with spinal muscular atrophy. Cell 80: 167-178, 1995

偍栤崌偣愭丗帠柋嬊丂丂![]() 丂
丂