文献
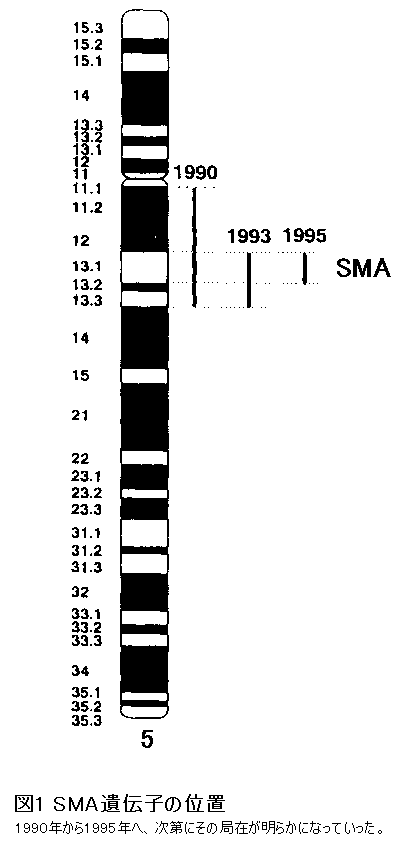
1) Brzustowicz LM, Lehner T, Castilla LH, et al. Genetic mapping of chronic childhood-onset spinal muscular atrophy to chromosome 5q11. 2-13. 3. Nature 1990; 344: 540-1.
2) Melki J, Abdelhak S, Sheth P, et al. Gene for chronic proximal spinal muscular atrophies maps to chromosome 5q. Nature 1990; 344: 767-8.
3) Kleyn PW, Wang CH, Lien LL, et al. Construction of a yeast artificial chromosome contig spanning the spinal muscular atrophy disease gene region. Proc Natl Acad Sci USA 1993; 90: 6801-5.
4) MacKenzie A, Roy N, Besner A, et al. Genetic linkage analysis of Canadian spinal muscular atrophy kindreds using flanking microsatellite 5q13 polymorphisms. Hum Genet 1993; 90: 501-4.
5) Melki J, Lefebvre S, Burglen L, et al. De novo and inherited deletions of the 5q13 region in spinal muscular atrophy. Science 1994; 264: 1474-7.
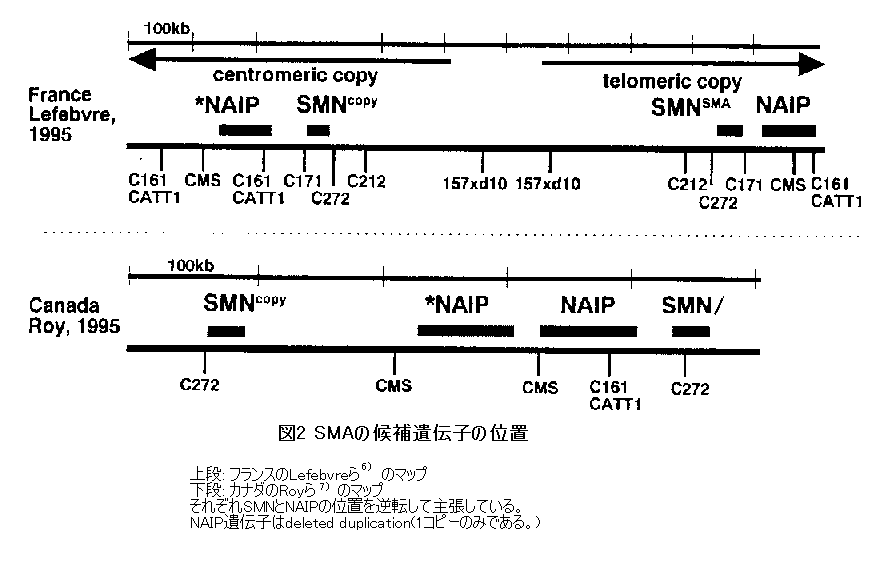
6) Lefebvre S, Burglen L, Reboullet S, et al. Identification and characterization of a spinal muscular atrophy - determining gene. Cell 1995; 80: 155-165.
7) Roy N, Mahadevan MS, McLean M, et al. The gene for neuronal apoptosis inhibitory protein is partially deleted individuals with spinal muscular atrophy. Cell 1995; 80: 167-178.
8) Wirth B, Hahnen E, Morgan K, et al. Allelic association and deletions in autosomal recessive proximal spinal muscular atrophy: association of marker genotype with disease severity and candidate cDNAs. Hum Mol Genet 1995; 4: 1273-84.
9) Velasco E, Valero C, Valero A, et al. Molecular analysis of the SMN and NAIP genes in Spanish spinal muscular atrophy (SMA) families and correlation between number of copies of cBCD541 and SMA phenotype. HumMol Genet 1996; 5: 257-63.
10) Rodrigues NR, Owen N, Talbot K, et al. Gene deletions in spinal muscular atrophy. J Med Genet 1996; 33: 93-6.
11) Lefebvre S, Burlet P, Liu Q, et al. Correlation between severity and SMN protein level in spinal muscular atrophy. Nature Genet 1997; 16: 265-9.
12) Munsat TL. Workshop report. International SMA Collaboration. Neuromusc Disord 1991; 1: 81.
13) Dubowitz V. Chaos in the classification of SMA: a possible resolution. Neuromusc Disord 1995; 5: 3-5.
14) van der Steege G, Groorscholten PM, van der Vlies, et al. PCR-based DNA test to confirm clinical diagnosis of autosomal recessive spinal muscular atrophy. Lancet 1995; 345: 985-6.
15) Shiraiwa Y, Saito K, Ikeda J, et al. Clinical feature and molecular genetic diagnosis of spinal muscular atrophy. J Tokyo Women's Medical College (in press).
16) DiDonato CJ, Ingraham SE, Mendell JR, et al. Deletion and conversion in spinal muscular atrophy patients: Is there a relationship to severity? Ann Neurol 1997; 41: 230-7
17) Burglen L, Seroz T, Miniou P, et al. The gene encoding p44, a subunit of the transcription factor TFIIH, is involved in large-scale deletions associated with Werdnig-Hoffmann disease. Am J Hum Genet 1997; 60: 72-9.
18) Liu Q & Dreyfuss G. A novel nuclear structure containing the survival of motor neurons protein. EMBO J 1996; 15: 3555-65.
19) Rudnik-Schoneborn S, Forkert R, Hahnen E, et al. Clinical spectrum and diagnostic criteria of infantile spinal muscular atrophy: Further delineation on the basis of SMN gene deletion findings. Neuropediatrics 1996; 27: 8-15.
20) Kausch K, Muller CR, Grimm T, et al. No evidence for linkage of autosomal dominant proximal spinal muscular atrophies to chromosome 5qmarkers. Hum Genet 1991; 86: 317-8.
21) Melki J, Burlet P, Clemont O, et al. Refined linkage map of the chromosome 5 in the region of the spinal muscular atrophy gene. Genomics 1994; 15: 521-4.
22) Melki J, Lefebvre S, Burglen L, et al. De novo and inherited deletions of the 5q13 region in spinal muscular atrophies. Science 1994; 264: 1474-7.
23) DiDonato CJ, Morgan K, Carpten JD, et al. Association between Ag1-CA alleles and severity of autosomal recessive proximal spinal muscular atrophy. Am J Hum Genet 1994; 55: 1218-29.
24) Francis MJ, Morrison KM, Campbell L, et al. A contig of non-chimeric YACs containing the spinal muscular atrophy gene in 5q13. Hum Mol Genet 1993; 2: 1161-7.
25) Wirth B, El-Agwany A, Burghes A, et al. Mapping of the spinal muscular atrophy (SMA) gene to a 750kb interval and the identification of linkage disequilibrium in the German population. Eur J Hum Genet 1995; 3: 56-60.
26) Wang CH, Kleyn PW, Vitale E, et al. Refinement of the spinal muscular atrophy locus by genetic and physical mapping. 1995; 56: 202-9.
27) Wirth B, Rudnik-Schoneborn S, Hahnen E, et al. Prenatal prediction in families with autosomal recessive proximal spinal muscular atrophy (5q11.2-q13.3): molecular genetics and clinical experience in 109 cases. Prenatal Diag 1995; 56: 202-9